
Clinical trials are the engine behind every new treatment we see – from life-saving cancer drugs to vaccines that help us fight global pandemics. But behind the scenes, running these trials involves juggling massive amounts of complex, sensitive data.
For pharmaceutical companies, outdated technology systems can slow everything down, delaying access to critical therapies. That’s why there’s a growing need to modernize how clinical trial data is collected, managed and analyzed.
In this post, we outline a modern clinical tech platform architecture that combines principles from general modern data platforms with the necessity of validated environments for regulated workflows. This architecture facilitates the efficient and compliant management, analysis and reporting of clinical trial data.
Whether you’re a clinical data programmer, informatics architect or therapeutic development leader, or simply curious about how new medicines come to life, this breakdown shows how smarter tech can lead to faster cures.
Why modernization matters in clinical trials
The pharmaceutical trial sponsor faces significant challenges in managing the increasing volume and diversity of data. Outdated systems, often characterized by fragmented, siloed data and cumbersome point-to-point connections, hinder innovation and delay treatments. Modernizing the clinical trial development tech stack is essential to overcoming these obstacles and accelerating the development of life-saving therapies.
Universal data platforms such as Databricks, Snowflake or other lake house cloud storage technologies offer advantages in integrating diverse data types and providing a comprehensive view of patient health. However, a validated “compliance-first” Statistical Computing Environment (SCE) remains critical for highly regulated clinical trial processes leading to submissions.
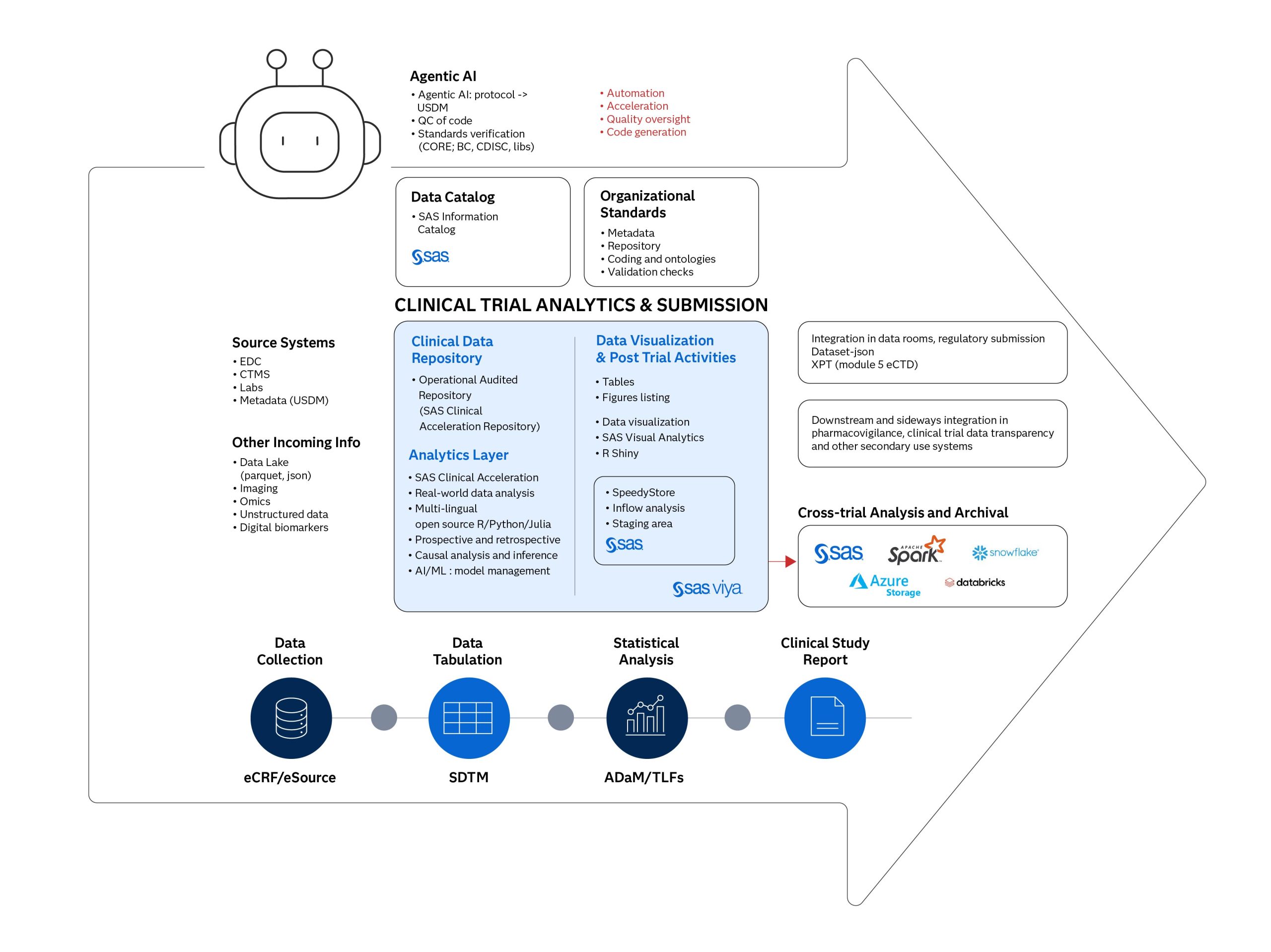
The clinical data repository: The foundation of trusted data
A clinical data repository (CDR) or data layer is at the core of this modern architecture. This repository is designed to integrate and manage diverse clinical data from various sources, including EDC, labs, EMR/RWD, imaging data and omics data. Universal platforms built on flexible architectures, such as cloud object storage, can be well-suited for generic data storage but not for CDRs, as they enable clinical data storage at scale and handle all data types, including unstructured formats such as those derived from digital biomarkers.
The CDR, however, serves as a central source of truth, ensuring data integrity and collaboration. Essential features for regulatory compliance include robust data governance and security, incorporating role-based access control, user traceability, secure storage, audit trails, electronic signatures, versioning and adherence to regulations like FDA Title 21 CFR Part 11 and compliance with data standards. The data pedigree should be traced back to the source data. Clinical data repositories such as SAS Clinical Acceleration Repository can provide robust governance tools and capabilities like lineage tracking, audit trails and access control to support these requirements within the CDR.
Turning data into regulatory-ready insights
Building on this governed data foundation is the analytics layer, which serves as the environment for analysis and insight generation. This layer needs robust architecture and high-performance and scalable computing capabilities to handle concurrent usage, reliable calculations after the last patient visit timepoints during trial conduct and provide insights through advanced analytics and machine learning for modelling and simulation purposes. It must support core statistical analysis, data exploration and integration of different programming languages such as SAS, R and Python.
This analytics environment must be validated and GxP compliant for regulated workflows and submissions, ensuring data integrity, security and reliability. This environment is crucial for generating the analysis results required for regulatory submissions, such as Tables, Listings and Graphs (TLGs). Reproducibility and transparency are vital goals supported by a strong SCE, which provides an environment to document workflows, share code and ensure consistency across different studies.
Trusted, validated platforms power the core of regulated clinical trials
While universal data platforms provide a strong foundation for a generic data store and can handle various data processing tasks, the architecture acknowledges the need for dedicated, validated environments for the final stages of regulated processes, such as producing analysis datasets (ADaM) and TLGs for submission. SAS has been an integral part of the pharmaceutical industry for many years, offering proven, industry-leading analysis software and a robust team of support engineers.
SAS® Viya® is highlighted as a cloud-enabled analytics platform designed to handle complex data and provide insights through advanced analytics and machine learning. SAS provides validated SCEs built for GxP compliance, ensuring data integrity, security and reliability through meticulous planning, validation and ongoing maintenance. SAS’ Clinical Acceleration Repository supports validated environments, data integrity and collaboration. These SAS components operate within a framework of robust data governance and security, including a metadata repository holding crucial information like mappings and standards and orchestration/workflows manage the data flow and support CI/CD practices.
Related: Drive speed to market with more efficient clinical trials
For regulated submissions, the emphasis remains on the validated, controlled environment offered by platforms like SAS. The capabilities of SAS Viya are described as reproducible, traceable, auditable, precise, secure, collaborative and GxP-compliant.
While open-source programming languages like R and Python are valuable for certain analytical tasks, our experience in real-world deployments at most pharmaceutical sponsors indicates that for the core workflows of clinical data management and statistical analysis in regulated trials, established platforms independent of language choice often serve as the primary, trusted backbone.
Relying solely on general-purpose open-source tools for these critical functions, or attempting to replace a proven, domain-specific platform entirely with them, can be akin to changing lanes in a traffic jam on a crucial journey; it overlooks the specific, rigorous demands of the clinical trial process, including stringent requirements for reproducibility and auditability and ignores the need to innovate by applying new technologies and capabilities.
As highlighted by industry analysis, achieving benefits such as faster study start-up, accelerated trial execution, higher productivity and increased success rates requires a strategic approach that prioritizes validated, integrated capabilities to truly modernize and accelerate clinical trials.
Powering the future of clinical trials with agentic AI and metadata
Beyond core analysis, the future involves leveraging technologies like agentic AI to enhance efficiency. Agentic AI is characterized by context awareness, complex reasoning, and problem-solving capabilities. AI agents, or multi-agent frameworks, can automate and accelerate complex analytical tasks and workflows.
As shown conceptually in the diagram, these agents can work in teams with specialized expertise (like data engineers, statisticians, or governance managers) to decompose complex tasks, plan steps, query data, write code and even critique results.
Integrating AI capabilities within a validated environment could streamline processes from data retrieval and analysis to report generation, advancing the continuum from exploratory insights to reproducible regulatory outputs.
This requires a scalable framework with configurable and testable agents, workgroups and workflows, governance guardrails, security, compliance rules, monitoring, version control and audit logs.
Furthermore, end-to-end metadata management, often aligned with CDISC standards, is fundamental for driving automation and ensuring traceability and reproducibility. Concepts like CDISC 360 are crucial for linking source data, transformations, analysis datasets (like SDTM and ADaM), and reports through a robust metadata layer. CDISC standards are essential, including CDASH for CRFs, SDTM for tabulation data, ADaM for analysis data, Define-XML for metadata and Controlled Terminology.
Automating processes like SDTM mapping driven by metadata and global data standards can lead to significant efficiency gains. This end-to-end metadata flow, managed effectively within the platform, enables automated checks, supports data tracing back to the source and ensures data integrity throughout the clinical data lifecycle.
By combining modern data management principles and a robust purpose-built CDR such as SAS Clinical Acceleration Repository, comprehensive governance and security, a qualified and compliance-first analytics environment (such as those provided by SAS Viya) for regulated workflows and the strategic integration of advanced technologies like agentic AI and end-to-end metadata management aligned with standards like CDISC, organizations can build a flexible, scalable and compliant platform capable of accelerating clinical trials and bringing new therapies to patients faster.



